 La Fibrodisplasia Osificante Progresiva (FOP) conocida también por: miositis osificante progresiva, enfermedad de Münchmeyer, enfermedad del hombre de piedra, es un desorden hereditario del tejido blando. Esta rara enfermedad genética en la cual el cuerpo produce "hueso adicional" en areas donde no debe existir. Este “hueso adicional” se desarrolla en el interior de músculos, tendones, ligamentos y otros tejidos conectivos.
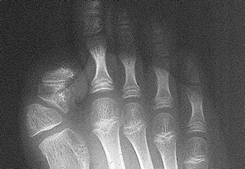
La Fibrodisplasia Osificante Progresiva (FOP) conocida también por: miositis osificante progresiva, enfermedad de Münchmeyer, enfermedad del hombre de piedra, es un desorden hereditario del tejido blando. Esta rara enfermedad genética en la cual el cuerpo produce "hueso adicional" en areas donde no debe existir. Este “hueso adicional” se desarrolla en el interior de músculos, tendones, ligamentos y otros tejidos conectivos.Una pista valiosa para su diagnóstico correcto es un dedo gordo del pie corto, independientemente de si el dedo pulgar es corto o no, la FOP es la principal causa de hallux valgus congénito.
Esta enfermedad afecta a 2.500 personas en todo el mundo (a uno de cada dos millones.) En España puede haber aproximadamente 18 enfermos de fop AEFOP
En este enlace podréis leer un estudio realizado a finales del 2014 por
Antonio Morales-Piga del Instituto de Investigación de Enfermedades Raras, Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), Instituto de Salud Carlos III, Madrid, España, Francisco Javier Bachiller-Corral del Servicio de Reumatología, Hospital Ramón y Cajal, Madrid, España, Gonzalo Sánchez-Duffhues del Departamento de Biología Molecular y Celular del Leids Universitair Medisch Centrum (LUMC), Leiden, Holandac
En el presente artículo, resumen los últimos avances con implicaciones
que trascienden los límites de esta devastadora enfermedad para
postularse como un nuevo modelo dentro de la fisiopatología humana.
En conclusión comprender en profundidad la mutación asociada a la FOP, la ruta de
señalización de las BMP y el mecanismo de «transición
endotelial-mesenquimal» no solo proporciona nuevas dianas terapéuticas
para obtener fármacos eficaces frente a esta enfermedad, sino que supone
un esperanzador avance en el conocimiento de una variedad de procesos
básicos en la metamorfosis y la reparación de tejidos y órganos.

No hay comentarios:
Publicar un comentario